Overview
The Ha laboratory is interested in studying the role of genomic alterations in cancer progression and translating this knowledge to expand applications for precision medicine.
We combine research in two complementary areas:
- Develop and apply novel computational methods to comprehensively profile cancer genomes from tumor tissue in large patient cohorts.
- Develop approaches to exploit liquid biopsies, such as circulating cell-free DNA from blood plasma, for studying cancer.
We leverage insights from the analysis of tumor genomes to inform the design of blood-based applications to monitor patient response to treatment. Our goals are to uncover mechanisms of treatment resistance, to identify blood-based genetic biomarkers, and to translate these findings to help improve clinical decisions. We are actively engaged in collaborations with research and clinical colleagues, including at Fred Hutchinson Cancer Center and UW Medicine to study different tumor types.
1. Liquid Biopsies to Study Cancer
from cancer patients.
Liquid biopsies, such as blood plasma containing cell-free DNA (cfDNA), provide a non-invasive route to study cancer. In cancer patients, cfDNA can contain circulating tumor DNA (ctDNA) released from dying tumor cells. ctDNA is more accessible than tumor tissue, particularly in advanced stage cancers when tumor biopsies are difficult to obtain. We are interested in the framework of using ctDNA for enhancing the study of large cancer patient cohorts that have less accessible tumors. The strategy of studying both tumor and cfDNA sequencing will help to establish a new paradigm in large-scale cancer genome studies.
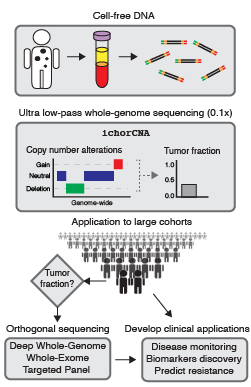
Estimating ctDNA content using cost-effective approach
Our lab develops novel computational approaches and pipelines for high-sensitivity copy number and tissue-of-origin analysis from the sequencing of cfDNA. We developed the tool, ichorCNA, which estimates the tumor fraction and copy number alterations from cost-effective ultra-low pass genome sequencing (Adalsteinsson*, Ha*, Freeman* et al. Nat Commun, 2017).
Tumor phenotype analysis from cell-free DNA
Current research and clinical efforts have focused on mutation detection in select cancer genes from ctDNA. However, approaches to characterize the tumor phenotype from ctDNA is still a nascent area of research and development.
Recently, we developed Griffin, which is a framework for profiling nucleosome accessibility from cell-free DNA for studying transcriptional regulation and tumor phenotypes (Doebley et al. Nat Commun, 2022). We have applied these to classify breast cancer hormone status (Doebley et al. Nat Commun, 2022) To study the transcriptional activity from ctDNA, we developed TritonNP to capture nucleosome patterns associated with active transcription in key regulators of prostate cancer (De Sarkar* and Patton* et al. Cancer Discovery, 2022).
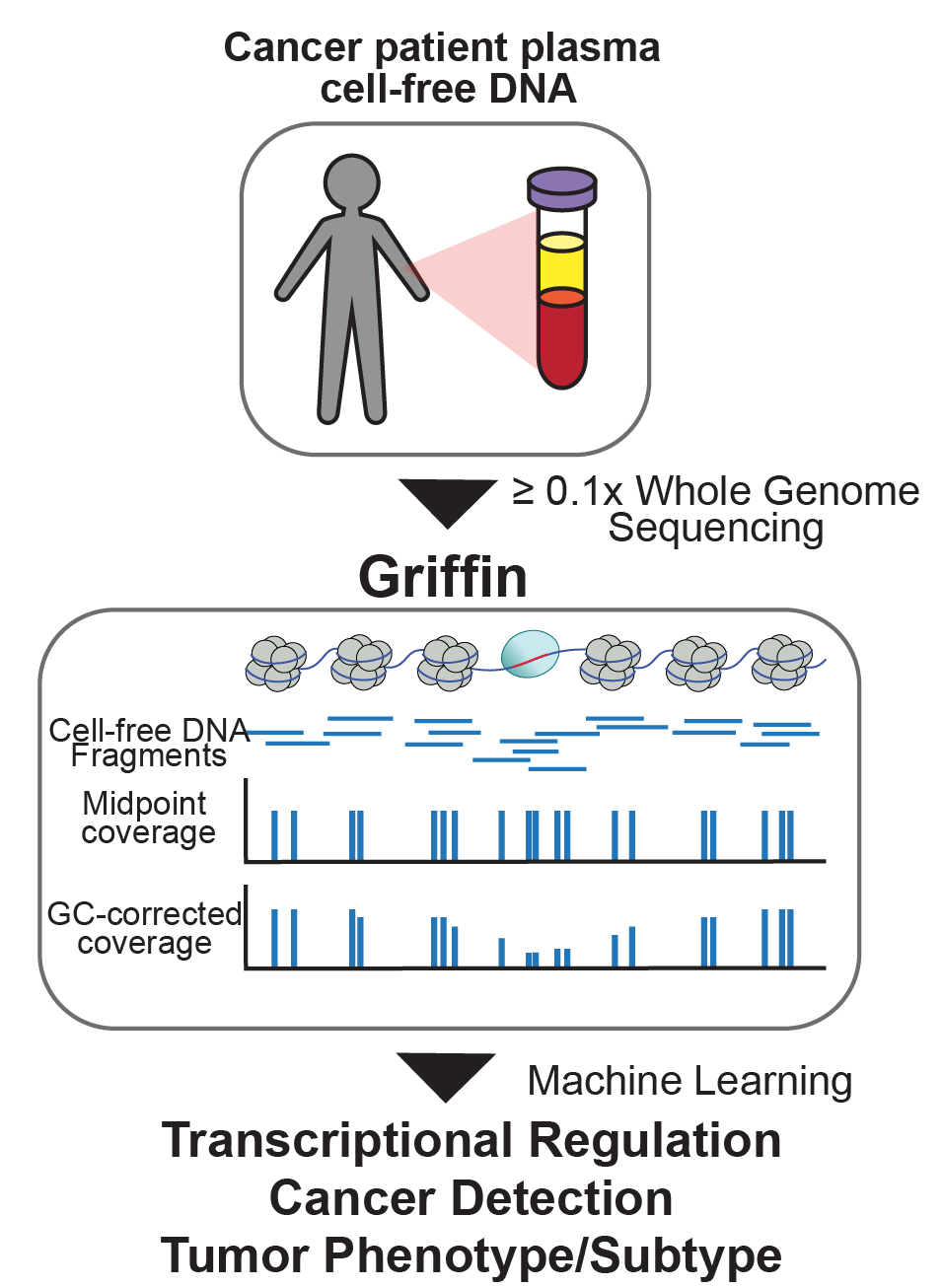
cancer detection and tumor subtyping.
(Doebley et al. Nat Commun, 2022; Fig 1)
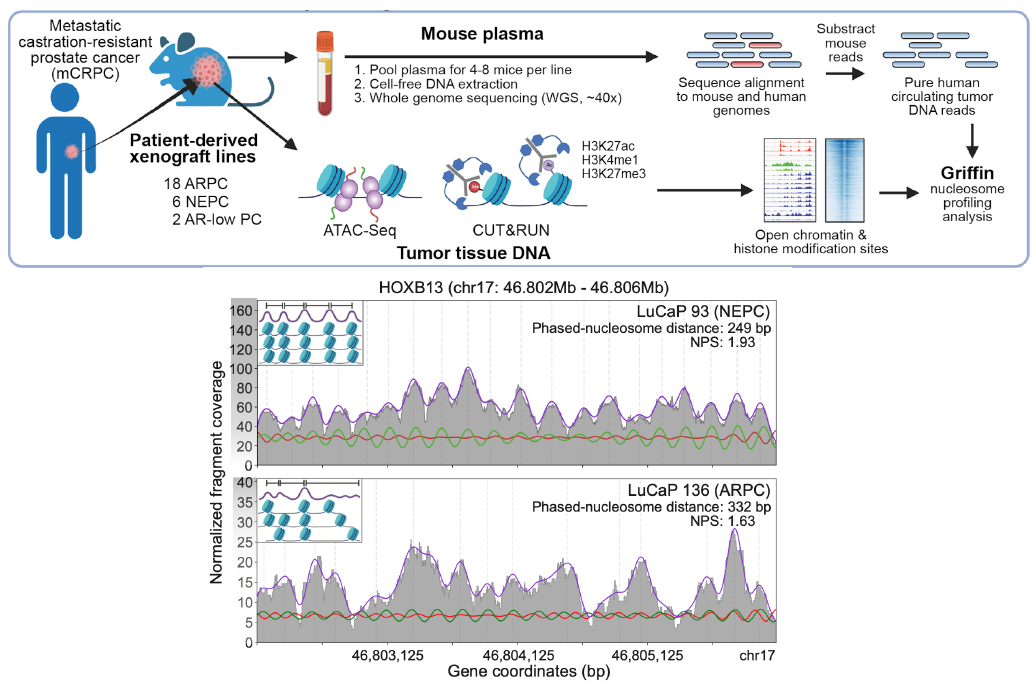
(De Sarkar*, Patton* et al. Cancer Discovery, 2022; Fig 1A and 3A)
2. Development of Methods for Precision Oncology
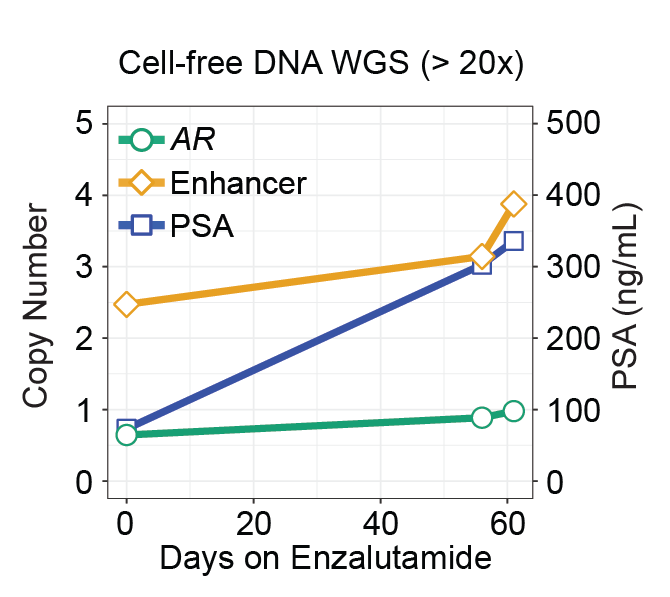
(Viswanathan*, Ha*, Hoff* et al. Cell, 2018; Fig 7C)
A challenge in precision medicine is the ability to routinely sequence patients’ tumors because repeated biopsies are not feasible, especially for advanced cancers. Liquid biopsy is an attractive solution for monitoring tumor genomic alterations from circulating tumor DNA (ctDNA). However, the systematic analysis to detect genome-wide signatures from ctDNA is still needed to understand the feasibility of longitudinally monitoring the dynamics of tumor clones in circulation. The lack of robust informatics solutions is a major barrier to realizing the potential of cfDNA. The development of sensitive approaches for analyzing ctDNA are needed to further strengthen the utility of cfDNA for cancer genome diagnostics, identification of biomarkers, and patient monitoring of response to therapy.
Monitoring genomic alterations from blood during therapy
We are developing novel algorithms to detect and validate tumor-specific alterations in ctDNA and to use these aberrant genomic events as reliably markers to monitor patient response to treatment. By increasing the sensitivity for detecting alterations in ctDNA, we will work towards important applications for clinical tracking of cancer burden in patients, perhaps even early stage cancer detection.
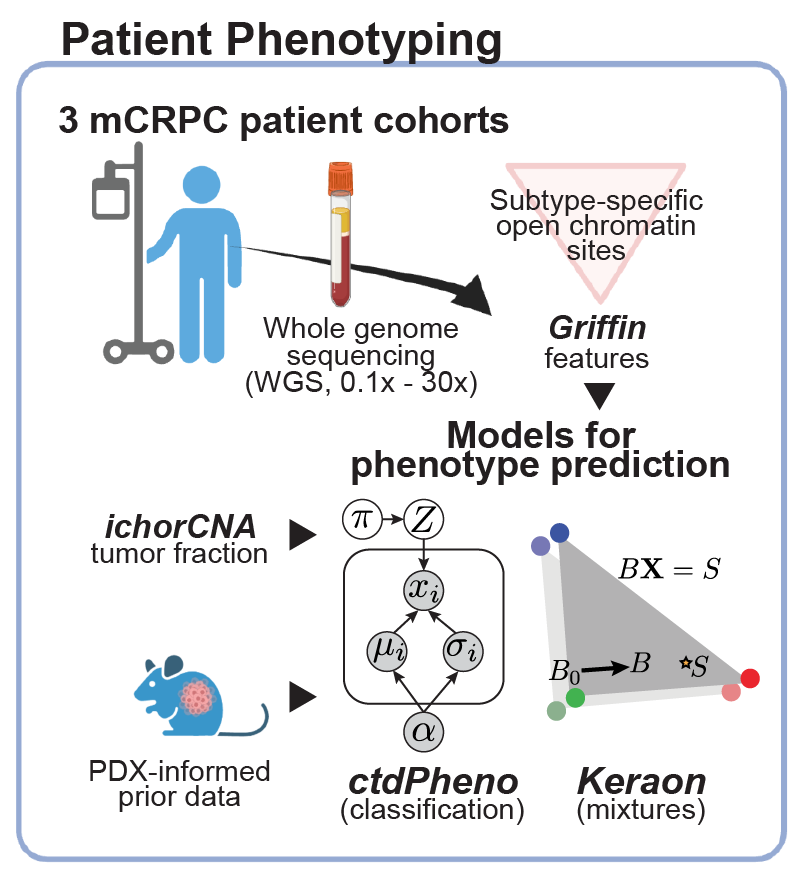
prediction in patient plasma samples.
(De Sarkar*, Patton* et al. Cancer Discovery, 2022; Fig 1)
Classifying tumor subtypes and phenotypes in patient plasma
Our group has developed new computational methods for classifying subtypes or phenotypes tumors from ctDNA, such as distinguishing adenocarinoma and neuroendocrine protate tumors and breast cancer hormone subtype. We developed ctdPheno, which uses low-coverage sequencing of ctDNA, and Keraon, which can also estimate the proportions of each phenotype De Sarkar* and Patton* et al. Cancer Discovery, 2022). We have open projects to apply these methods on new patient cohorts to test their utility for predicting response to standard-of-care and novel therapeutics.
3. Application of New Technologies to Study Genomic Alterations in Tumor Genomes
Analysis of whole genome sequencing (WGS) of tumor genomes can uncover genome-wide alterations, including structural rearrangements and mutations in non-coding features, which are not easily interrogated by whole exome sequencing (WES). Genomic rearrangements can alter non-coding features, including the duplication of enhancers of oncogenes and relocation (‘hijacking’) of enhancers to proto-oncogenes. The reconstruction of these genome rearrangements will be important to understanding the their role in cancer.
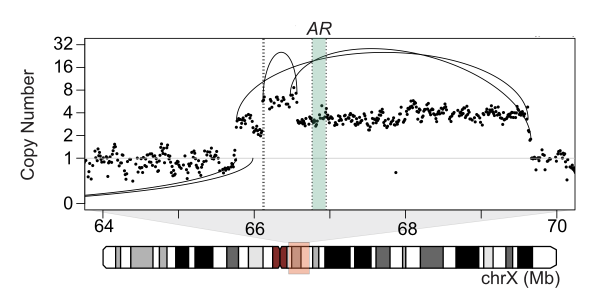
(Viswanathan*, Ha*, Hoff* et al. Cell, 2018; Fig 4A).
Our lab is interested in studying the abnormal structure of the cancer genome more deeply. We apply cutting-edge whole genome DNA sequencing technologies, particularly platforms that generate long-range genomic information such as linked-read and long-read data. These data enhance the reconstruction of genomic rearrangements and enable the study of alterations in non-coding genomic regions. We are also interested in integrating chromosome conformation information to better understand the effects of genomic alterations on the 3D chromosome structure.
Our work in advanced prostate and breast cancers, as well as work by others, have revealed non-coding structural rearrangements altering enhancer elements and driving expression of cancer genes. There is a need for more comprehensive analyses of cancer genomes to uncover new non-coding alterations driving cancer progression. We are interesting in performing whole genome characterization of metastatic cancers, from both tumor and liquid biopsies, to address major research questions, such as treatment resistance.
4. Development of Computational Methods for Cancer Genomics
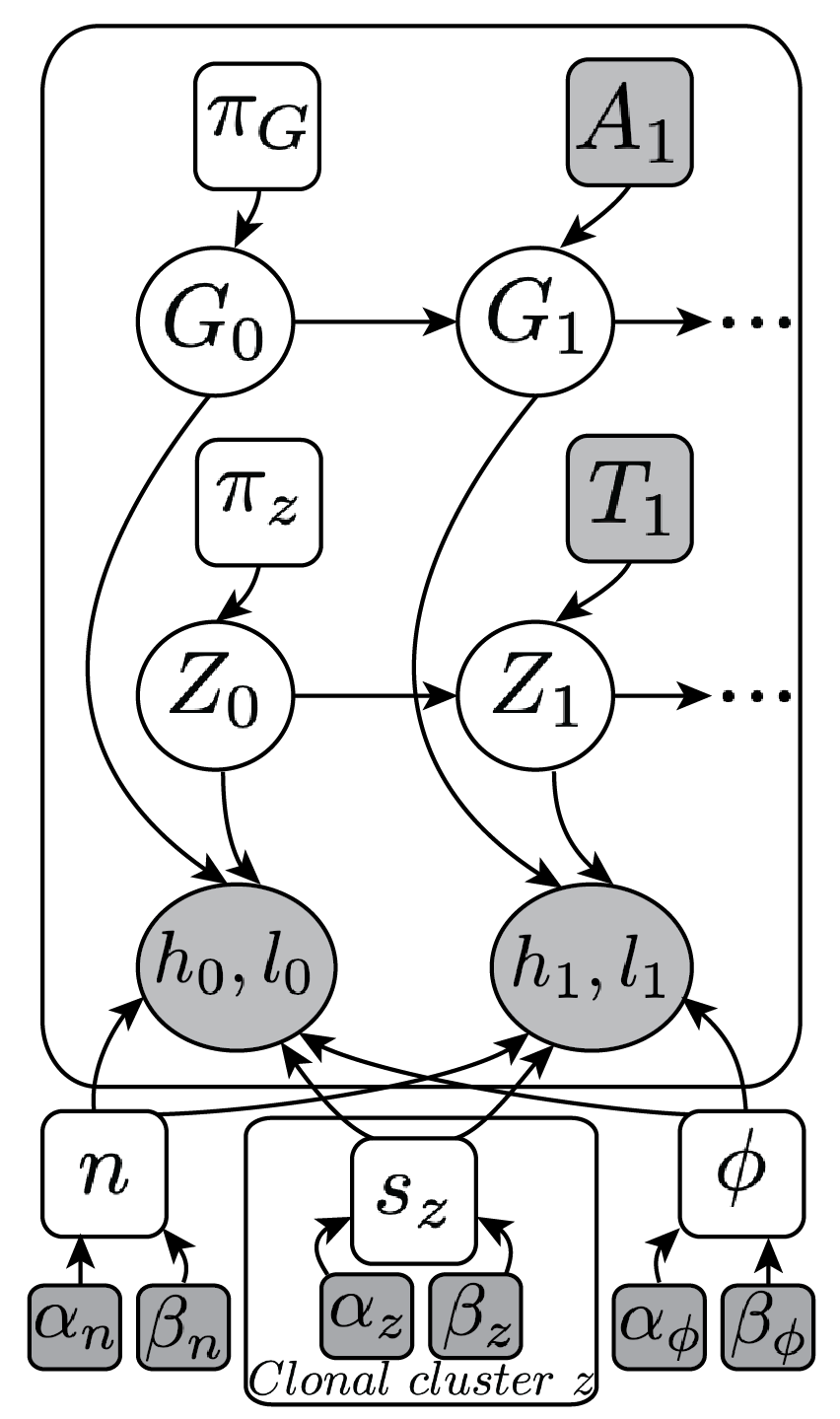
(Ha et al. Genome Res, 2014)
We develop tools that employ machine learning algorithms to analyze cancer genomes. We use statistical/probabilistic models to develop novel algorithms and frameworks for predicting the aberrant structure of rearranged genomes in cancer. These include tools such as TITAN, which predicts subclonal copy number in tumors with intra-tumor heterogeneity.
Current genome sequencing approaches have limited capabilities for interrogating low complexity, repetitive non-coding regions and for accurate reconstruction of complex rearrangements. The advent of new experimental technologies to generate long-range genomic information, such as linked-read sequencing (10X Genomics), is beginning to address these deficiencies. However, these approaches are still in their infancy and lack robust analytical tools to decipher these data and to realize their potential for understanding cancer genome structure. We have extended or modified current algorithms to leverage these new data features to improve prediction of copy number alterations and structural rearrangements (see Software).
Funding
The Gavin Ha Lab is generously supported by funding from the following institutes, foundations, and agencies. Dr. Ha is also supported by the following awards:
- NIH Director’s New Innovative Award (DP2 CA280624)
- Transition Career Development Award (NCI, K22 CA237746),
- Prostate Cancer Foundation Young Investigator Award (PCF YIA)
- V Scholar Grant from The V Foundation
